
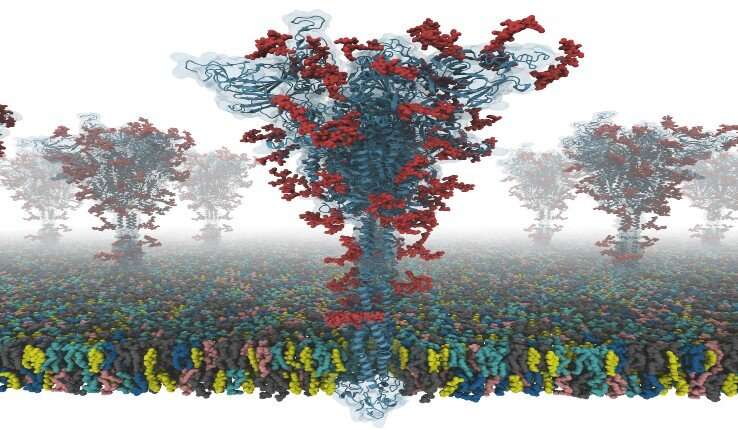
Das virus SARS-coronavirus 2 (SARS-CoV-2) ist die bekannte Ursache der coronavirus Krankheit 2019 (COVID-19). Der „spike“ oder S-protein erleichtert den Eintritt von Viren in Wirtszellen.
Nun hat eine Gruppe von Forschern von der Lehigh und der Seoul National University in Südkorea und der University of Cambridge in Großbritannien haben gemeinsam produzieren die erste open-source all-atom Modelle eine volle-Länge S-protein. Die Forscher sagen, dies ist von besonderer Bedeutung, da die S-protein spielt eine zentrale Rolle in der viralen Eintritt in die Zellen, so dass es ein Ziel für Impfstoff und antiviralen Medikamenten-Entwicklung.
Dieses video zeigt, wie über den Bau der Membran-system, aus Ihrer SARS-CoV-2 S-protein-Modelle. Das Modell-Gebäude-Programm ist open access und kann von der Homepage des CHARMM-GUI auf der COVID-19-Archiv.
Entwickelt von Wonpil Im, professor an der Lehigh der Abteilung von Biologischen Wissenschaften und Bioengineering Department, CHARMM-GUI (graphical user interface) ist ein Programm, das simuliert komplexe biomolekulare Systeme, die einfach, präzise und schnell. Im beschreibt es als „computational Mikroskop“, das ermöglicht es den Wissenschaftlern zu verstehen auf molekularer Ebene die Interaktionen, die nicht eingehalten werden können, keine andere Möglichkeit.
https://www.youtube.com/embed/1EhOWs-GZvQ?color=white
„Unsere Modelle sind die ersten voll-glykosylierten in voller Länge SARS-CoV-2 spike (S) protein-Modelle, die verfügbar sind, um andere Wissenschaftler“, sagt Im. „Ich hatte das Glück, gemeinsam mit Dr. Chaok Seok von der Seoul National University in Korea und Dr. Tristan Croll von der University of Cambridge in Großbritannien Unser team verbrachte die Tage und Nächte, um diese Modelle sehr sorgfältig von den bekannten cryo-EM Struktur Portionen. Die Modellierung war sehr anspruchsvoll, denn es gab viele Regionen, in denen einfache Modellierung versäumt, Modelle von hoher Qualität.“
Wissenschaftler können mithilfe der Modelle führen innovative und neuartige Simulations-Forschung für die Prävention und Behandlung von COVID-19, nach Im.
Die S-Struktur des proteins wurde bestimmt mit der cryo-EM mit der RBD-bis (PDB-ID: 6VSB), und mit der RBD unten (PDB-ID: 6VXX). Aber dieses Modell hat viele fehlende Rückstände. Also, Sie zunächst modelliert, die fehlenden Aminosäurereste, und dann den fehlenden Domänen. Zusätzlich, Sie modelliert alle möglichen Glykane (oder Kohlenhydrate), die an die S-protein. Die Glykane verhindern, dass Antikörper, die Anerkennung, die es schwierig macht, einen Impfstoff zu entwickeln. Sie Bauten auch eine virale Membran-system des S-proteins für die Molekulardynamik-simulation.